

In the vast realm of medical conditions, there are some that strike with a profound impact, both on the individuals they affect and the scientific community working to understand and combat them. One such condition is spinal muscular atrophy (SMA). Often referred to as a “silent killer,” SMA is a rare genetic disorder that affects the motor neurons responsible for controlling muscle movement. In this article, we will embark on a journey to uncover the mysteries surrounding spinal muscular atrophy, delving into its causes, symptoms, and ongoing efforts to find effective treatments.
Unraveling the Genetic Threads
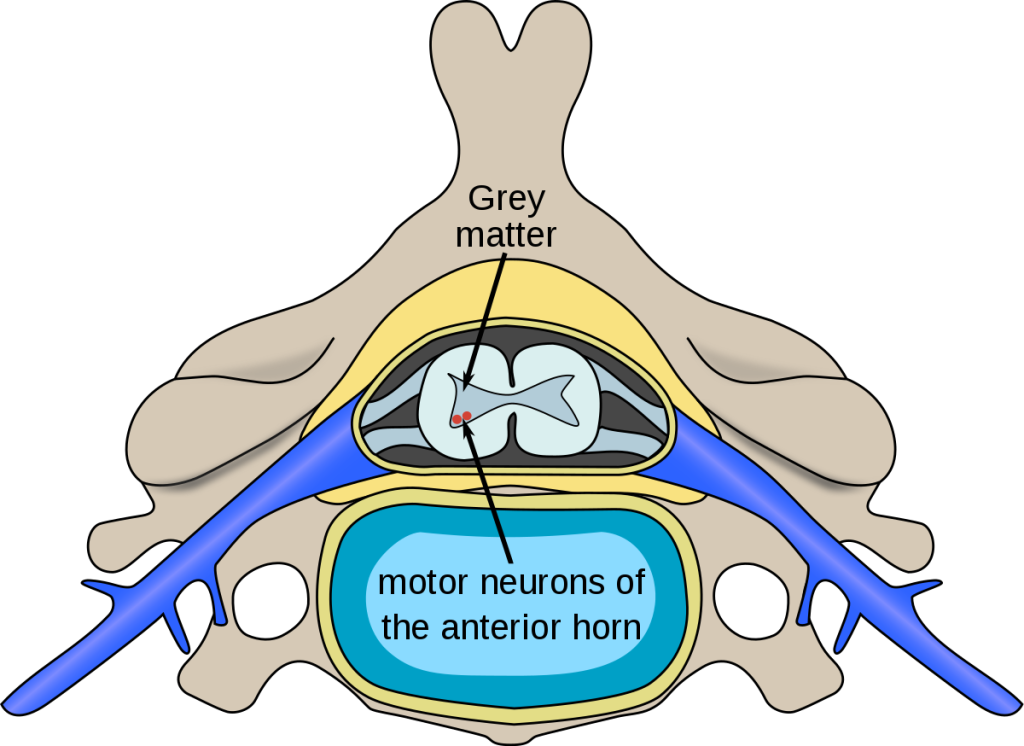
At the core of spinal muscular atrophy lies its genetic nature. SMA is primarily caused by a mutation in the survival motor neuron 1 (SMN1) gene, which plays a crucial role in the production of survival motor neuron (SMN) protein. Without sufficient SMN protein, motor neurons in the spinal cord progressively degenerate, leading to muscle weakness and atrophy. However, it is important to note that SMA can also result from mutations in other genes, such as the neuronal apoptosis inhibitory protein (NAIP) gene.
The severity of SMA can vary significantly, classified into different types based on the age of onset and the progression of symptoms. The most severe form is known as SMA type 1, typically appearing in infants and leading to severe muscle weakness and respiratory difficulties. On the other end of the spectrum is SMA type 4, which usually manifests later in life and presents with milder symptoms. Understanding these genetic intricacies allows researchers to develop targeted approaches to diagnose and treat SMA.
Detecting the Signs
Early detection plays a crucial role in managing spinal muscular atrophy. Recognizing the subtle signs can lead to timely interventions and improved outcomes for individuals living with SMA. Symptoms may vary depending on the type and stage of the condition, but common indicators include muscle weakness, poor muscle tone, difficulty breathing, and delayed motor milestones in infants.
Diagnostic tools such as genetic testing and electromyography (EMG) are vital in confirming a suspected SMA diagnosis. Genetic testing can identify mutations in the SMN1 gene, providing valuable insights into the severity and progression of the disease. Meanwhile, EMG measures the electrical activity of muscles and can help differentiate SMA from other neuromuscular disorders. Early detection empowers medical professionals to develop personalized treatment plans and support individuals with SMA to live fulfilling lives.
Navigating the Path to Hope
Although spinal muscular atrophy poses significant challenges, the scientific community has made tremendous strides in recent years. One of the breakthroughs in SMA research is the development of disease-modifying therapies that target the underlying genetic causes. One such treatment is a medication called nusinersen (brand name Spinraza), which is administered through a spinal injection. Nusinersen works by increasing the production of SMN protein, thus slowing the progression of SMA.

Another promising avenue in SMA research is gene therapy. Gene replacement therapies like onasemnogene abeparvovec (brand name Zolgensma) offer hope by providing a functional copy of the SMN1 gene to individuals with SMA. This groundbreaking approach has shown remarkable results, particularly in infants with SMA type 1, significantly improving survival rates and motor function.
Conclusion
Spinal muscular atrophy is a complex and challenging condition that affects individuals and families worldwide. By understanding the genetic basis of SMA, recognizing its early signs, and exploring innovative treatments, we can move closer to unraveling its mysteries and offering hope to those affected. With continued research and advancements, the journey to conquer spinal muscular atrophy moves forward, illuminating a path of progress and possibilities for a brighter future.